The purpose of developing this multiresidue method was to determine low part per billion levels of four active ingredients and eight metabolites in soil. The soil samples were obtained from a field soil dissipation study in which the active ingredients were applied in the order of one gram of active ingredient per hectare. The use of these low application rates in the field work of the seed treatment use pattern necessitated an order of magnitude improvement in sensitivity from standard LC/UV, GC/MS, and LC/MS analytical methods. In addition, the number and diversity in polarity of these analytes could not be analyzed in a single procedure as was done here. The objectives that were considered in the method development phase involved the use of an efficient extraction solution, an appropriate concentration and clean-up procedure, and sensitive and selective analytical instrumentation. Once these objectives were obtained, the scope of the study became the generation of Good Laboratory Practices (GLP) compliant data using a reliable and rugged method of analysis for use in soil dissipation studies.
The classifications of the four parent pesticides of interest were a triazine
insecticide (thiamethoxam), an acylalanine fungicide (metalaxyl M), a triazole
fungicide (difenoconazole), and a pyrrole fungicide (fludioxonil).
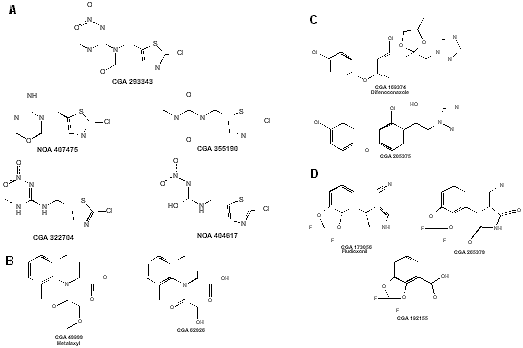
Figure 1. Chemical structures of residues of concern: (A) thiamethoxam and metabolites, (B) metalaxyl M and
metabolite, (C) difenoconazole and metabolite, and (D) fludioxonil and metabolites.
Each sample was extracted by shaking with an acidified acetonitrile-water mixture. The sample was then sonicated and centrifuged followed by the removal of an aliquot and its dilution with acidified water. The diluted aliquot was cleaned-up on a styrene divinylbenzene co-polymer (ENV) and a strong cation exchange (SCX) Solid Phase Extraction (SPE) cartridge connected in tandem. The analytes were eluted from each cartridge separately and analyzed by a Triple Quadrupole Mass Spectrometer.
All the quantitative analytical reference substances were obtained from Novartis Crop Protection, Inc., Greensboro, NC.
Preparation of the soil samples was done by blending with solid carbon dioxide in a Hobart food cutter to obtain an uniform granular powder. The moisture content was determined gravimetrically.
A 25 g subsample of wet soil was weighed into a 250 mL centrifuge bottle and the fortification samples were spiked. After the fortification the sample bottles were capped and the contents mixed manually. The cap was then removed and the sample was allowed to stand for a minimum of 5 minutes. 75 mL of the extraction solution (1:24:75 acetic acid:water:acetonitrile (v/v)) was added to each sample. The samples were shaken by hand for 10 15 seconds and then for a minimum of 30 minutes on a mechanical shaker. The sample was sonicated for 10 15 minutes and centrifuged at 5000 6000 rpm for approximately 10 minutes. The supernatant was decanted through a funnel fitted with a cotton ball plug into a 250 mL graduated cylinder. The extraction was repeated once more and the combined extracts brought to 150 mL with 1% acetic acid in water (v/v). A 25 mL aliquot was removed and transferred to a 500 mL Erlenmeyer flask containing 475 mL of 1% acetic acid in water. The solution was mixed and any particulate matter was allowed to settle before applying to the SPE cartridges.
A 1000 mg Varian ENV SPE cartridge was conditioned with 15 mL of methanol followed by 15 mL of 1% acetic acid in water on a vacuum manifold. The liquid level was not allowed to drain below the top of the sorbent bed at any point in this clean-up procedure. A 75 mL reservoir with adapter was connected to the top of the conditioned ENV cartridge. A 1000 mg Varian SCX SPE cartridge was conditioned with 10 mL of hexane followed by 10 mL of methanol. The conditioned ENV cartridge, with the attached reservoir, was connected to the top of the conditioned SCX cartridge with an adapter. The diluted sample aliquot was passed through the cartridges with a vacuum rate of approximately 10 mL per minute. The reservoir and cartridges were washed with 10 mL of deionized water. The SCX cartridge was detached and placed on a separate vacuum manifold. 65 mL of methanol was added and the analytes were eluted from the ENV cartridge at 1 - 2 drops per second. The eluent was transferred with methanol for concentration. The SCX cartridge was washed with 5 mL of methanol and then a 75 mL reservoir was connected to the SCX cartridge with an adapter. NOA 407475 was eluted from the SCX cartridge with 30 mL of methanol saturated with potassium chloride at a rate of one drop every 2 seconds. The elution solution was stirred 10 minutes before use to allow any undissolved potassium chloride to settle. The eluent was transferred to a 125 mL round bottom flask with methanol. The ENV eluent was evaporated to less than 1 mL using nitrogen evaporation with a 35 40 oC water bath and then brought to 1 mL in methanol. The round bottom flask containing the SCX eluent was placed on a vacuum rotary evaporator with a water bath temperature of approximately 40 oC. The extract was concentrated to less than 1 mL, transferred, and diluted to 2 mL with methanol.
The ENV extract was diluted 1:5 with acetonitrile and all the extracts were stored in a freezer at approximately 20 oC. The SCX extract was diluted 1:10 immediately after its removal from the freezer. The transfer of any precipitated potassium chloride crystals was avoided.
An automated Waters 715 Ultra WISP sample processor (Millipore, Milford, MA) was used for the injection of sample extracts and a model 9012 ternary solvent delivery system (Varian, Walnut Creek, CA) was used for the pumping of the mobile phases. A 4.6 cm by 25 cm (5 m particle size) Supelcosil octadecyl chromatography column (Supelco, Bellefonte, PA) with a flow of 0.8 mL/min. was used in the reverse phase. An isocratic liquid chromatograph profile was run at 25% 0.1 mol/L acetic acid in water and 75% acetonitrile. The analysis of NOA 407475 used a 4.6 cm by 15 cm (5 m particle size) Zorbax SCX chromatography column (Hewlett-Packard, Orangeville, ON) with a flow rate of 1.5 mL/min. of 50% 25 mmol/L ammonium acetate in water and 50% acetonitrile. An injection volume of 100 L was used for the analysis of NOA 407475 and 50 L for the analysis of all other analytes.
The analysis was carried out using a Perkin Elmer Sciex Atmospheric Pressure Ionization (API) Biomolecular Mass Analyzer (PE Sciex, Concord, ON). The sample introduction and ionization to the mass spectrometer interface was done in two ways; Electrospray Ionization (ESI) using a Turbo Ion Spray (TIS) inlet and Atmospheric Pressure Chemical Ionization (APCI) using a Heated Nebulizer (HN) inlet. Both inlets used nitrogen at 60 psi for the nebulizer gas pressure, 1.2 L/min. for the curtain gas flow, and 4.5 L/min. and 5 L/min. for the auxiliary gas flow for the TIS and HN inlets, respectively. The TIS inlet was operated at 450 oC and the HN inlet at 500 oC. The collision gas thickness was approximately 2.5x1015 argon molecules per cm2. The post column eluent was split 5:1 for use on the TIS inlet and 3:1 on the HN inlet for the analysis of NOA 407475 only.
Table 1 lists the operating parameters for each ionization technique and the ion mode of analysis. The instrument conditions were found to be optimal for the instrument used, however, the values must be optimized for each instrument. Table 2 lists the mass transition(s) for each analyte and the corresponding type of analysis.
Negative ion MS/MS used the molecular anions as the precursor ions and
positive ion MS/MS used the protonated molecular ions as precursor ions.
However, the precursor ion for CGA 62826 was a protonated, doubly hydrated
molecular ion. Product ions were formed by Collision Activated Dissociation
(CAD) of the precursor ions in the collision cell of the instrument. The
predominant product ions were mass analyzed in the third quadrupole.
Table 1. MS parameters for the analysis using the TIS and HN inlets for all four injections.
| Parameter | TIS | HN |
| Positive Ion Mode | Negative Ion Mode | Positive Ion Mode #1 | Positive Ion Mode #2 |
| Period 1 | Period 2 | Period 1 | Period 2 | Period 1 | Period 1 | |
| Ionization Voltage | 3500 V | 4000 V | - | - | - | - |
|
Discharge Current |
- | - | - 5 A | - 5 A | 5 A | 5 A |
| Orifice Potential | 60 V | 70 V | - 40 V | - 69 V | 49 V | 49 V |
| Initial Ion Energy | 3 V | 3 V | - 0.5 V | - 3 V | 4 V | 4 V |
| Collision Energy | 16 V | 26 V | - 18 V | - 30 V | 11 V | 11 V |
| Dwell Time | 100 ms | 200 ms | 250 ms | 250 ms | 100 ms | 250 ms |
Table 2. MS/MS transitions for each analyte and their corresponding analytical technique.
| Parent | Analyte | Mass Transition | Inlet / Ion Mode |
| Compound Group | (m/z m/z) |
| Quantitation | Confirmation | |||
| A
(CGA 293343) |
CGA 293343 | 292.2 210.8 | 292.0 180.8 | HN, Positive Ion #1 |
| CGA 322704 | 249.8 168.8 | - | ||
| CGA 355190 | 247.8 174.8 | - | ||
| NOA 404617 | 237.0 175.0 | - | TIS, Positive Ion, Period 1 | |
| NOA 407475 | 247.0 160.8 | - | HN, Positive Ion #2 | |
| B
(Metalaxyl) |
CGA 48988 | 280.1 220.1 | 280.1 248.1
280.1 192.1 280.1 160.1 |
HN, Positive Ion #1 |
| CGA 62826 | 288.2 113.0 | - | TIS, Positive Ion, Period 1 | |
| C
(Difenoconazole) |
CGA 169374 | 405.9 251.0 | 407.9 253.0 | TIS, Positive Ion, Period 2 |
| CGA 205375 | 350.0 70.0 | 352.0 70.0 | ||
| D
(Fludioxonil) |
CGA 173506 | 246.9 180.0 | 246.9 169.0
246.9 126.0 |
HN, Negative Ion, Period 2 |
| CGA 192155 | 201.0 90.9 | - | HN, Negative Ion, Period 1 | |
| CGA 265378 | 277.1 210.9 | 277.1 182.9 |
The instrument conditions were found to be optimal for the instrument used, however, the values must be optimized for each instrument.
Negative ion MS/MS used the molecular anions as the precursor ions and positive ion MS/MS used the protonated molecular ions as precursor ions. However, the precursor ion for CGA 62826 was a protonated, doubly hydrated molecular ion. Product ions were formed by Collision Activated Dissociation (CAD) of the precursor ions in the collision cell of the instrument. The predominant product ions were mass analyzed in the third quadrupole.
An external standard method of quantitation was performed using weighted (1/x)
linear regression calibrated over a four to five point calibration plot. The
calculations of residue recoveries were accomplished by the use of Perkin Elmer
Sciex MacQuan 1.5 quantitation software. Table 3 illustrates the recovery
statistics and detection limits for all the analyte candidates in this validation.
Fortification samples were done in duplicate at 3, 10, and 25 ng/g, except for
CGA 62826 and NOA 404617 which were done at 9, 30, 75 ng/g and NOA 407475 which was done at 15, 50, and 125 ng/g. The unfortified control soil
samples were found to be free of interferences.
Table 3. Statistical values for the recovery of fortification samples and the associated limit of detection.
|
Parent Compound Group |
Analyte | Mean Recovery | Standard Deviation | Relative Standard Deviation | Number of Fortification Samples | Limit of Detection |
| (%) | (%) | (%) | (ng/g) | |||
| A
(CGA 293343) |
CGA 293343 | 115 | 5.7 | 5.0 | 6 | 1.0 |
| CGA 322704 | 113 | 5.7 | 5.0 | 6 | 1.0 | |
| CGA 355190 | 112 | 2.7 | 2.4 | 6 | 1.0 | |
| NOA 404617 | 77 | 8.2 | 11 | 6 | 3.0 | |
| NOA 407475 | 68 | 5.5 | 8.1 | 6 | 2.0 | |
| B
(Metalaxyl) |
CGA 48988 | 110 | 6.5 | 5.9 | 6 | 1.0 |
| CGA 62826 | 156 | 44 | 28 | 6 | 3.0 | |
| C
(Difenoconazole) |
CGA 169374 | 90 | 13 | 14 | 5 | 1.0 |
| CGA 205375 | 105 | 38 | 36 | 5 | 1.0 | |
| D
(Fludioxonil) |
CGA 173506 | 102 | 4.5 | 4.4 | 6 | 0.30 |
| CGA 192155 | 113 | 11 | 9.7 | 6 | 1.0 | |
| CGA 265378 | 103 | 17 | 17 | 6 | 1.0 |
Due to a less consistent linear response of the hydrated CGA 62826 precursor ion recoveries of this analyte were more varied particularly at the 9 ng/g fortification level. Therefore, the results for this compound were considered to be only semiquantitative. Residue recovery results for CGA 205375 exhibited a standard deviation of + 38%. However, eliminating a recovery of 172% at the 3 ng/g fortification level the average recovery became 89% with a standard deviation of + 10% (RSD 11% for n = 4).
CGA 265378 underwent tautomerism in solutions that were primarily aqueous. The conversion products were observed at the same mass transition, but with less retention, which is consistent with an alcohol eluting on a nonpolar chromatography column (Figure 4). This conversion was reduced by using solutions having a high acetonitrile content.
For some precursor ions, matrix effects were observed and thus inhibited evaporation in the TIS inlet resulting in signal suppression. These effects, attributed to co-extractive organic compounds, were typically reduced by the use of the HN inlet. However, due to the thermal instability of the hydrated CGA 62826 precursor ion, only the TIS inlet was acceptable. There was no significant matrix effects in the analysis of CGA 169374, CGA 205375, and NOA 404617 by either inlet.
Also, due to the co-elution of analytes having different MS parameters separate analytical runs were required (Figures 3 - 6).
The procedure was based on a strategy of doing minimal sample processing and using the resolving power of the quadrupole magnets to achieve the specificity and sensitivity required. The simple extraction procedure using one process for all the residues of concern provide a great improvement in efficiency in the analysis. The excellent results show that this was achieved for most of the residues of concern. It is remarkable that a single procedure was possible, given the wide range of polarity and chemical properties of the residues of concern. Although this was not done, it is reasonable to expect that the use of isotopically labelled compounds as internal reference standards would eliminate the variability encountered with CGA 62826 and CGA 205375.
The advantages of the extraction phase of the procedure included the use and efficiency of a single extraction solution on such a diverse range of polarities. Employing SPE cartridges in the clean-up phase avoided the use of numerous and/or complicated liquid-liquid partition solvent systems for the extraction of chemistries ranging from acids to bases. The absence of derivatization reagents allowed for a simpler and less hazardous work-up.
The choice of a triple quadrupole mass spectrometer afforded many advantages over contemporary instrumentation. The sensitivity and resolution of the quadrupole provided reliable fingerprint identification of each residue of concern. The specificity of selected mass transitions ensured the absence of false positives and the selectivity minimized sample clean-up.
The extraction and analysis combined allowed for a high sample throughput.
The authors would like to acknowledge Narong Chamkasem of Novartis Crop Protection United States, Inc. for his excellent suggestions and research in the analysis thiamethoxam and metabolites in soil.